処方薬が私たちの手に渡るまで -ゼロからの開発、治験、発売―
新薬の開発というと、現在は新型コロナウイルスのワクチンを思い浮かべる方も多いかもしれません。このワクチン開発については事情が特殊なため、異例のスピードですすんでいます。しかし基本的には、新薬が開発されるまでには莫大な費用と、短くても10年程度の時間がかかるものです。
私たちが普段、医師から処方される薬は安全で効果が保証されています。そして、これらが国民皆保険制度によって安価に手に入るのは世界的に見ても一部でしか実現されていません。ED・AGA治療薬は国民皆保険制度の対象ではありませんが、処方薬として同じプロセスをたどります。
 基礎研究
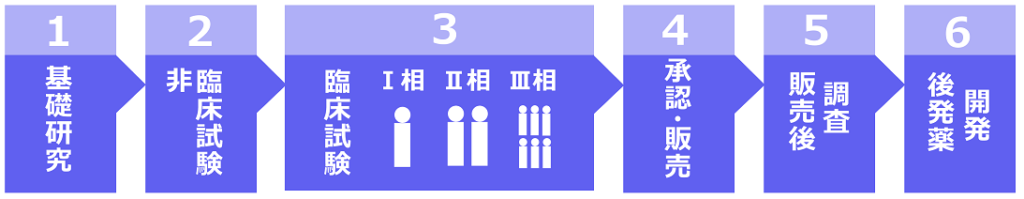
基礎研究
病気を生む要因を抑える可能性がある物質を探す。天然に存在する物質(植物・動物・鉱物など)からの抽出や、化学合成・バイオテクノロジーなどさまざまな技術を活用して、薬の候補となる化合物を作り、薬としての可能性を調べます。その候補を「リード化合物」と呼びますが、製薬会社が持つ、あるいは新たに持ってきた途方もない数の化学物質の中から、候補にかなうだけものを見つけ出す作業です。ほぼ全ての物質が対象ですから、気の遠くなるような過程です。候補となった新しい物質の化学構造を調べ、スクリーニング試験(新しい薬として有効な化合物を選択する作業)を繰り返し行い絞り込みます。
 非臨床試験
非臨床試験
基礎研究で発見した新しい物質の有効性と安全性を、動物や培養細胞を用いて確認します。人間でないものへの投与で、人間に与えた場合の予測を得るころが目的です。その物質が身体の中でどう広がり、どのような影響を与え、そして排泄されていくのかを観察します。その物質の好ましい作用や毒性、安定性に関する試験を行います。近年では動物愛護の観点から、動物実験の代替(培養細胞、コンピュータによるシミュレーションなど)、削減、改善(苦痛を和らげる方法の模索など)が積極的に行われています。
 臨床試験
臨床試験
非臨床試験を経た薬の候補が人体に有効で安全なものかどうかを調べる、いわゆる治験です。人体に適した用量や用法を見極めるために臨床試験は3段階に分かれており、ここで膨大な時間と費用を要することになります。病院などの医療機関で予め同意を得た健康な人や希望する患者を対象に、安全な投与量や投与方法などを確認する試験的な投与を行い、製品化された際の基準を決めてゆきます。- 第一相試験(フェーズI)
治験薬を少人数の健康な人で初めてテストします。少しずつ投与量を増やしながら、体内での吸収・排泄にかかる時間と、血液や尿などに含まれる薬の量を測定し、その安全性を確認します。
- 第二相試験(フェーズⅡ)
治験対象の患者の範囲をやや広げ、用量の範囲や適応する疾患の範囲を決定するために、効果があると予想される患者で治験薬の効果や副作用の程度を確認します。
- 第三相試験(フェーズⅢ)
治験薬を多数の患者を対象にテストを行い、前段階で予想した容量や使用法が適切かどうか、既存薬とも比較しながら実証を行います。
- 第一相試験(フェーズI)
 承認・販売
承認・販売
以上の試験を経て薬としての有効性・安全性が証明された後、厚生労働省への承認申請を行います。厚生労働省が医薬品医療機器総合機構(PMDA)に審査と、専門家などで構成する薬事・食品衛生審議会(薬事分科会)の審議を経て、厚生労働大臣の許可が下りると、医薬品として製造・販売することができるようになります。
 販売後調査(フェーズⅣ)
販売後調査(フェーズⅣ)
新しい薬の発売直後は使用数が急激にのび、承認前では予測できなかった副作用等に関する情報を早急に把握するため、「市販直後調査」を医薬品の製造販売業者が半年間実施します。
 処方薬の分類と、後発薬の開発
処方薬の分類と、後発薬の開発
こうした長い長いプロセスを経てやっと使えるようになった医薬品は、それらの管理がどの程度まで医療従事者に委ねられるべきかでいくつかに分類され(*処方薬と市販薬とのすみわけ)、一定のあいだ安全性が確認されると「後発薬」としてより安価な価格で使用することができるようになります。(*後発薬の開発(工事中))